In-house tools and pipelines
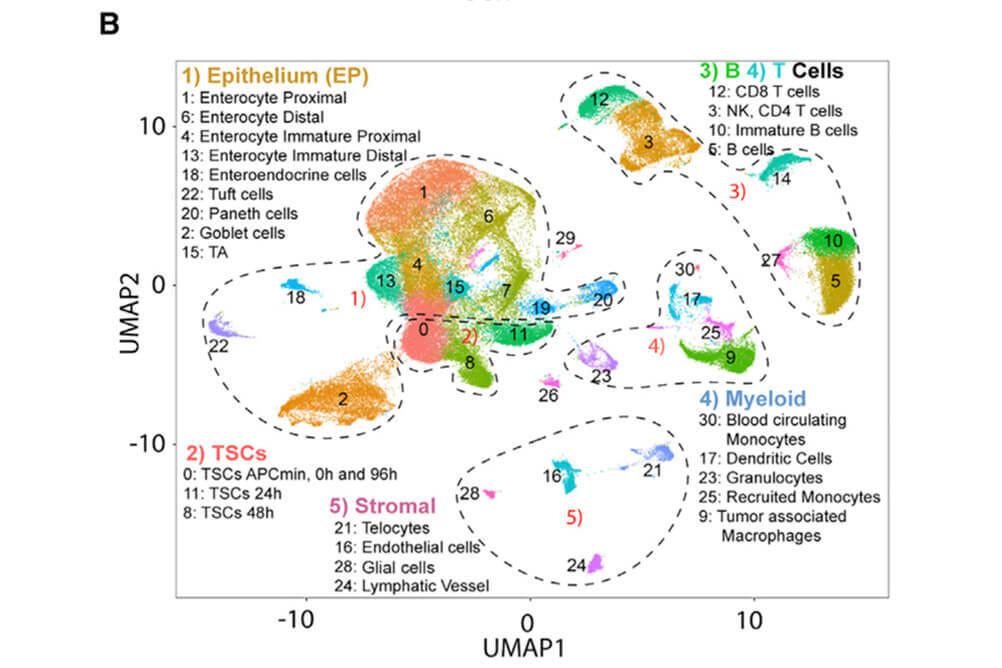
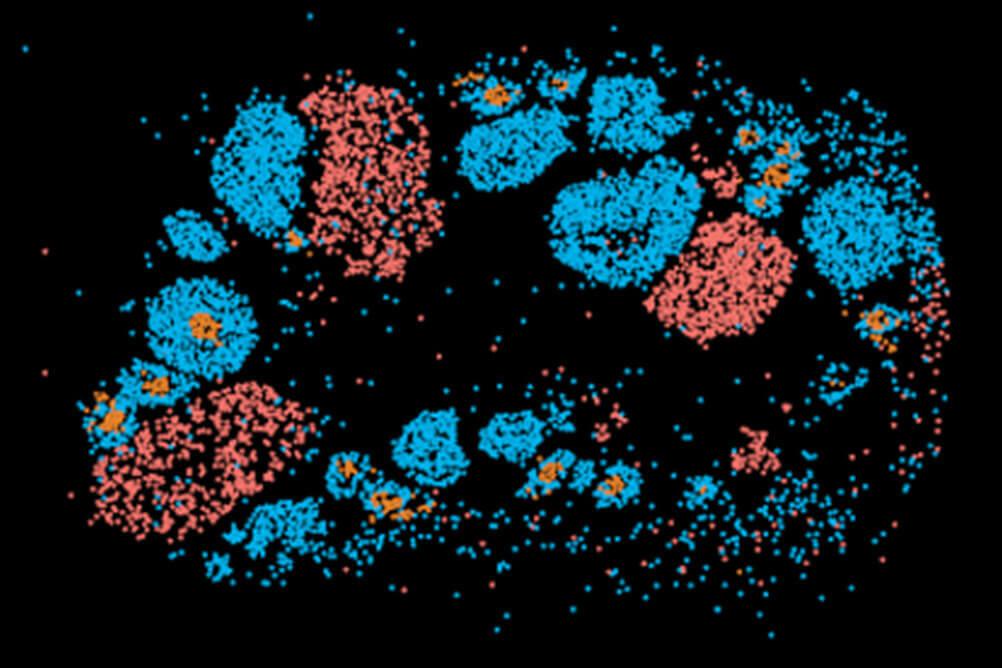
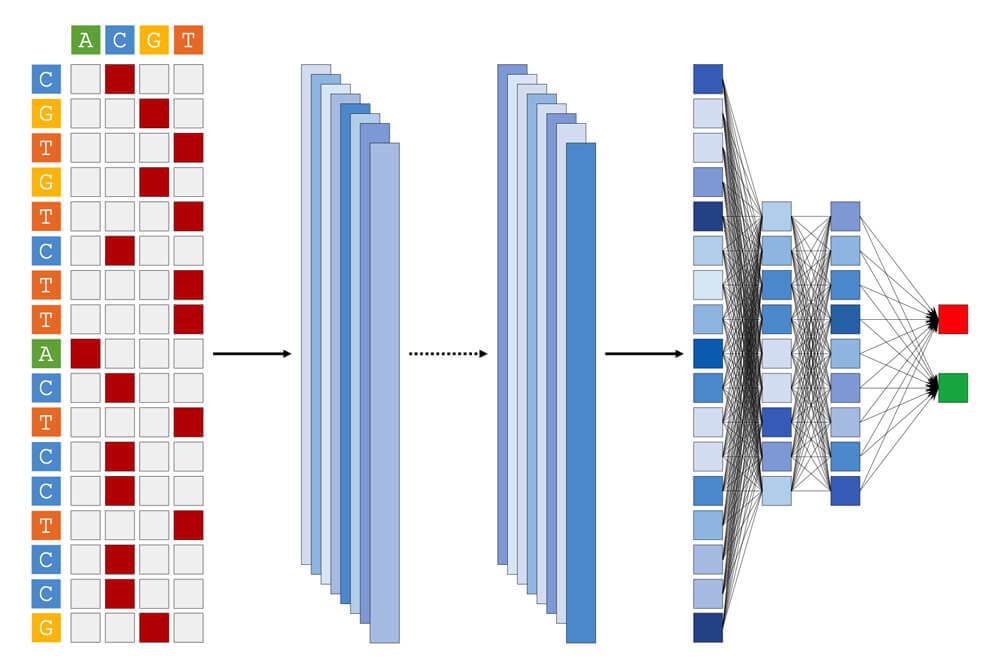
For data types we frequently encounter (RNA-seq, ChIP-seq, and single cell RNA-seq), we have developed robust pipelines to automatically run the first few steps of analysis and quality control. This saves time for challenging and more interesting downstream analyses, which varies from project to project.
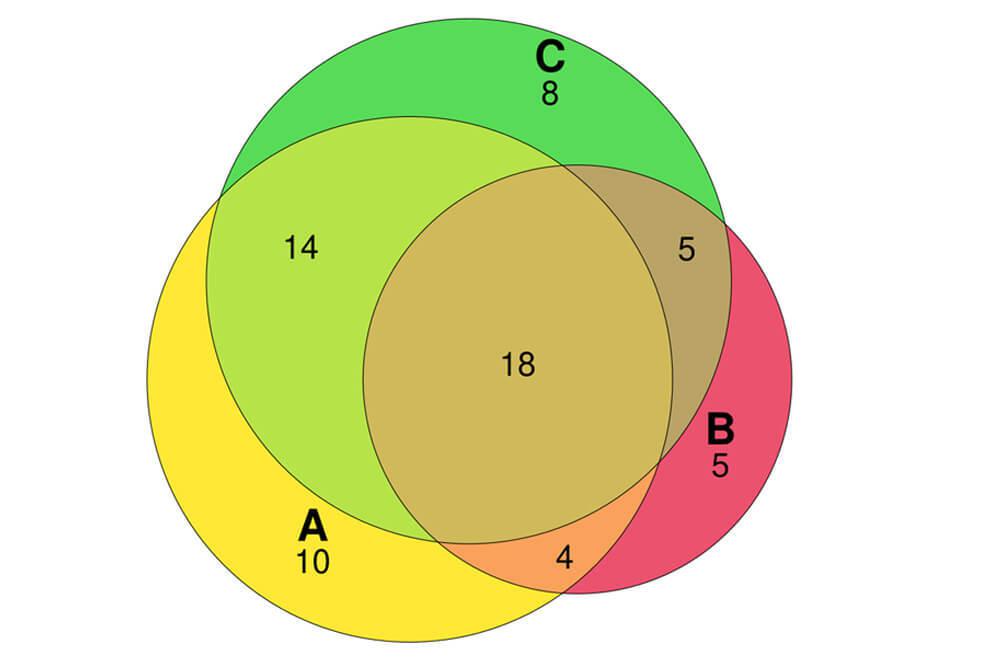
For aspects of analysis that we or our collaborators regularly perform, we have developed a number of in-house web applications – RNA-seq differential expression, gene ontology enrichment, Venn diagram construction, and sequencing depth needed for a given experiment. These tools enable institute members who don’t necessarily want to go through the process of learning programming to perform some basic analysis themselves.